The indestructible protein with no life and Genetic code: Havoc on the people of Fore - Papua New Guinea
It's been a while of absence from this lovely community. It is not without a good explanation; I simply went to seek more knowledge, and now I have some period of break from mental drilling, I am falling back to what I love doing, aside from seeking for knowledge.
Happy new year to you reading this and I trust your celebration went well. I must say, it's great to be back for a while. Winter break is upon us, and I have no other choice but to maximize it. Today's discussion bothers around a serendipitous discovery or will I say, a very interesting story and the scientific fact I stumbled on in my quest to prepare a PowerPoint presentation as a requirement for an exam.
Naturally, I love doing things differently from the conversational way, so I decided to surf the internet to find anything out of the normal. Right from my bachelor days, it has always been my mantra: look for the extraordinary, this way, you stay ahead of others and be different. I still approach my studies at the masters level with the same spirit. Interestingly, I stumbled on Transmissible Spongiform Encephalopathy, TSE. Whoops, don't let the long name scare you, we will sure scatter the big name as we progress. However, seen TSE was not the eureka moment for me because, it seemed more like every other brain disease. The actual YES! moment came when I discovered the unusual agent behind the disease. It is extraordinary I must say.
I believe you are now getting warmed up to know what this agent is, right? Alright, get some cup of coffee, adjust your position to a reading one and join me in this lovely interesting journey to the world of a pathogenic agent that does not have a life, no genetic code nor DNA/RNA but yet wrecks havoc in both humans and mammals. These agents are called Prions, an enemy within. Let's begin with a little but detailed history and introduction to this agent and why it is as dangerous as it can be when it finds its way to the brain.
Story time...
In the 1730s, there was a recorded incidence of degenerative diseases among sheep and goats. Little was known about this disease not until in 1838, when it was then first described. This disease notably affects the nervous system of this animals, thereby causing them to loose weight and always scratch their body against the wall. Amongst the common clinical signs also observed were changes in the bahavioural pattern, loss of their wools, itching, weird movement pattern amongst others. Of course, as a shepherd, you would sure notice these kind of changes easily, but the aetiology and cause of these problems remains the greatest challenge.
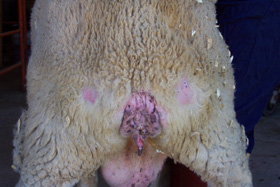
A sheep affected with Scrappie disease
{kind=link}
The disease was tagged incurable and most of the shepherds then were advised to stop inbreeding which obviously was discovered to be one of the propelling factor to the spread of the disease, in essence, the disease was contagious. In fact, people were advised to simply kill and dispose off the sheep once they notice the signs and symptoms of the disease in their sheep. By the year 1942, it was also observed in goats. It did not take long before the first case of this disease was identified in humans and was named Kuru. By 1970s and 80s, the true cause of this disease was later discovered to be cause by a pathogen called Proteinacious Infectious Particle, PRION. Now this is were it gets more interesting!
Researchers studying the disease then found that it was caused by a misfolded protein called a prion which is a normal protein found in the brain that can become abnormal and cause disease when it changes its shape. Even after discovery, though yet unnamed then, the scientists tried inactivating this agent with various pathogen inactivation techniques like boiling, use of some oxidizing agents that would on a normal day kill bacteria and viruses. All efforts proved abortive.
Two decades after a scientist named Gordon failed to inactivate the scrapie agent with formalin, other scientists began to investigate scrapie stability. Many experts from different fields tried to inactivate the scrapie agent by ionizing and UV irradiation, extreme heat, high pressures and other compounds known to inactivate viruses and bacteria
Obviously, this agent was indestructible. Upon carrying further studies and research, it became ultimately clear that prion was the real cause of the scrapie disease in the sheep and goats and in addition in even among the Fore people of Kuru in New Guinea. This disease appearance among Fore people, was the very first case of human transmission and thus was given the name Kuru, which in their native language means to tremble or shake. I am at this point really not so surprised because the name perfectly matches the signs their were observed in the infected individuals. It was discovered to affect the brains of the victims resulting to neurodegeneration, hence the reason for the uncoordinated movement, shaky and trembling features. Similarly, prion was also identified as the causative agent of this disease and There is a reason why this disease affected the Kuru people, read on!. In the meantime, let's take a further delve into this agent and why it seems indestructible by various means.
What are prions (Proteinacious Infectious Proteins?
Just like every other protein in the body, prions are normal proteins produced by the body and it is encoded by the gene - HRNP located on the short arm of chromosome 20. They are represented as PrP and can basically exist in two forms - a cellular or physiologic form, PrPc and also in a diseased or scrapie form, PrPsc. By nature, the normal type are found on cellular membranes of cells but research has shown that they mainly more abundant in the brain - the central nervous system and peripheral nervous system of mammals where they carry out physiologic functions. Till date, the well researched and known function of this prion protein is cellular signalling - myelination of the axon fibres in the brain and neuroprotection. Besides, there other documented functions of this protein.

Location of prion gene
{kind=link}
On the cell membranes (a structural layer on cells that separates them from the cytosol), you find them attached through a glycosylphosphatidylInositol, GPI anchor. However, they could under several cleavages in their alpha, Beta, Gamma regions and also another type of cleavage known as Shedding. You don't have to worry about this post translational modification (changes that occur on most proteins resulting to their full maturity and functionality), the most important thing you need to know here is that, these cleavages are very important for the full functionality of the protein so that it can break into fragments and carry out some physiological function. There is a reason why I had to go this deeper into the molecular structure of this protein.
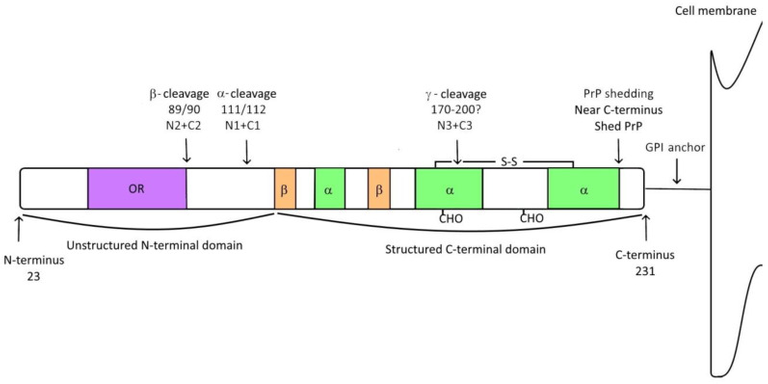
Molecular structure of the Prion protein
Take a look at the image above, along the amino acid sequence of this protein, there is a region known as the Octapeptide repeat region, OR (an area in a protein where you have repeated sequence of amino acids). This region of the protein according to research, is believed to be responsible for sporadic transformation of the normal PrPc into the abnormal type PrPsc. How this actually happens and why it happens is yet to be uncovered. In the normal conformation of this protein, it is arranged in an alpha helical manner that allows its flexibility but in diseased state, the case is different.
Normal prion proteins can sporadically undergo a transformational change into the diseased conformation, and when this happens, they change into the isoform of the protein that is highly rich in Beta Sheets. Remember, alpha helical conformation of most proteins confers flexibility, but when they assume a beta sheet conformation, they become more resistance to changes and even heat. Liken this to a cloth made from silk and the one made from cotton wool. Wool is made up of Alpha keratin, a very hard protein and hardly soluble in water. It has an alpha helix that is mainly stabilized by three types of bond but amongst, the disulfide bond which is contributed by cysteine (an amino acid that naturally has disulfide bridges) is the strongest.

Structure of the major prion protein in its normal alpha helical coformation
{kind=link}
Because of the alpha helical arrangement of the protein, when you steam clothes made from wool, you would notice that, the clothes simply squeezes and upon cooling, they hardly return back to their original appearance, unless you apply same heat i.e using steam irons. This flexibility and ability to return to their original conformation, is simply as a result of the alpha helical arrangement of the protein matrix. Now the case is different when you steam clothes made from silk. Silk on the other hand contains a type of protein called fibroin that are highly resistant to any form of mechanical stress or distortion.
Not necessarily because of the type of protein they have, but because of the type of conformation the protein fibers have. In the fibroin, the protein assume a Beta Sheet conformation thus giving it that great ability to withstand any form of mechanical stress in the form of heat. You can read more about the William Astbury experiment on fibrous proteins to learn more about this lovely discovery. The summary of the whole thing here is that, when protein assume a Beta conformation, they become more resistant to degradation and denaturing by heat or by proteases (enzymes that degrade proteins)
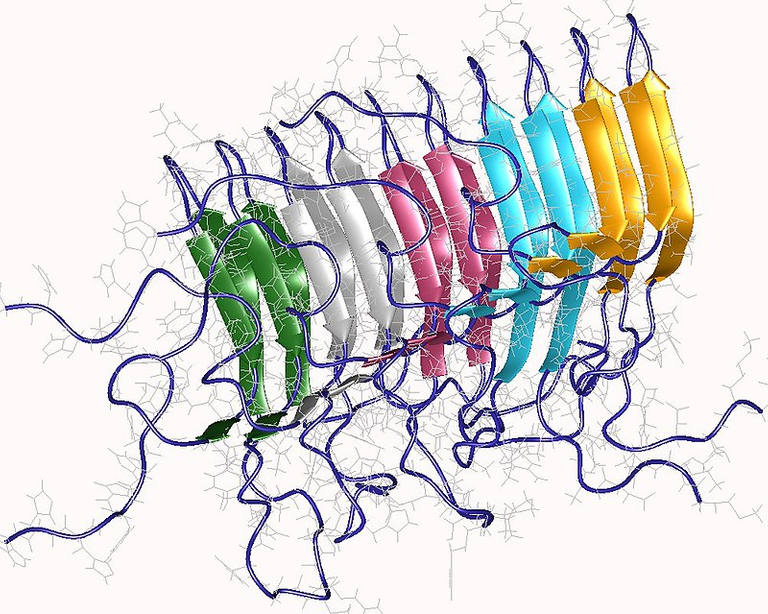
Beta-sheet rich amyloid conformation of Prion protein
{kind=link}
Now you know why this prion protein was unable to be inactivated in the early days of its discovery. Even till now, it is still very difficult to kill this agent. Well, you cannot possibly kill what is not alive. Are you surprise at this statement? Well read on, you will learn more about this protein. Some of the weird characteristics of the prion protein is that:
•They are infectious in nature
•They do not have any genetic code
•They are not alive, yet they infect other health alike proteins
•They do not die (Well they don't have life, so they can't die)
•They do not use energy
•They do not grow but they multiply
•The disease they cause is incurable
All the above characteristics makes the prion protein more deadly than we think. Diseases caused by Prion are collectively called Prion Diseases aka Transmissible Spongiform Encephalopathy, TSE. Breaking this down, by being transmissible, they can infect other healthy normal prion proteins in the brain, their causing them to change their normal alpha helical conformation to the Beta amyloid sheet conformation and the cycle continues. Spongiform simply explains the sponge like shape appearance of the brain cells infected and damaged by these diseased prions while the encephalopathy simply means that the disease is associated with the brain. It is expected, since the majority of this proteins are in the brain. Let's now look at the disease pattern of this destructive agent.
How prions operate and infect other proteins despite not having DNA nor RNA
Just like I earlier elucidated, prion proteins can be utterly devastating and infectious to other healthy ones upon contact. Their mode of infecting and causing normal prion proteins to assume a diseased state is a remarkable one.
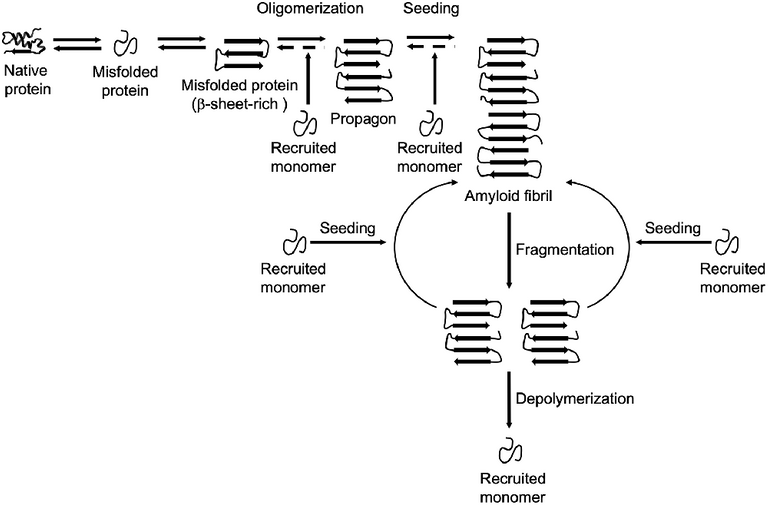
Amyloid formation and propagation at the molecular level. Native proteins oligomerize after adopting an abnormal β-sheet-rich fold, eventually forming a propagon. Propagons are specific units that can recruit and incorporate native monomers, which allows them to grow into amyloid fibrils. Fragmentation events can lead to complete depolymerization into monomers or to the formation of new propagons that in turn provide more ends for recruiting monomers.
Recall that we earlier explained that normal prions PrPc can sporadically undergo conformation change and assume an isoform that is highly autocatalytic in nature, PrPsc i.e it can continue to replicate on its own, spreading and infecting other healthy PrPc prions. The conversion from the healthy to diseased form is more like the conversion from a well folded protein to a misfolded protein. This misfolded proteins when in excess can then stack together to form what is known as amyloid fibrils ( a product of several folding of B-sheets fibers.
This Beta sheets later form a longer sheet known as the propagon and like the name suggest, its functions to propagate the diseased prions by recruiting more misfolded protein monomers. This continues like a chain reaction, thereby infecting more cells and causing them to change and assume a wrong conformation.
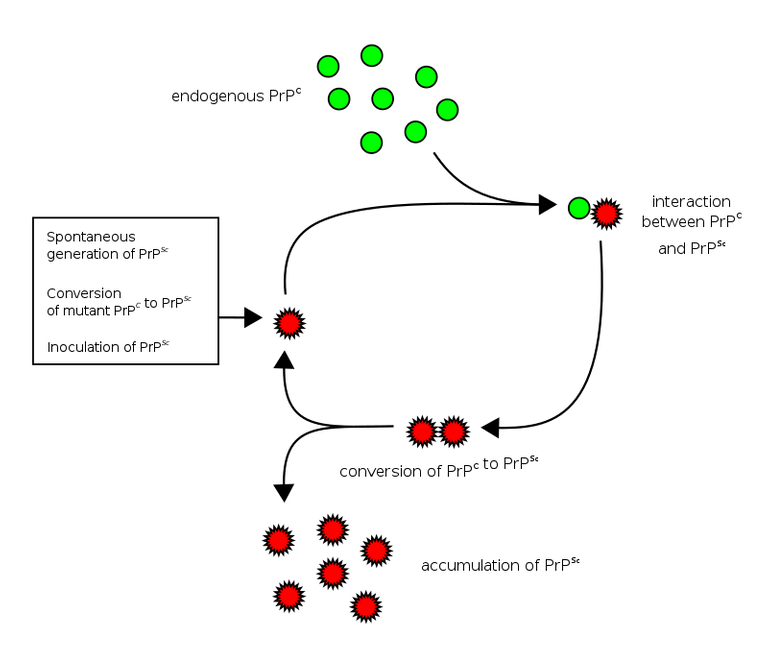
How prions infect other health prion protein
{kind=link}
Normal cellular prions are susceptible to degradations by proteases like proteasome which enzymatic ability. Note, in eukaryotic and prokaryotic cells, a molecule known as the Chaperone ensures that nascent and newly produced proteins fold properly with their hydrophobic site buried inside while exposing their hydrophilic region. For proteins to be active and functional, they must fold and fold correctly and chaperones do this job effectively. In situations where these proteins do not fold correctly, it can cause destructive effects and deposits.
Before the formation of B-sheet rich conformation, chaperones can still convert the misfolded protein back to its original normal conformation or they are degraded by the proteasomes, but when this fails, which in most cases fail, the resultant effect is an excess accumulation of Amyloid fibrils of the diseased PrPsc prion protein in the brain of the patient. Depending on who is affected, the disease is so named. In humans, they cause Creutzfeldt–Jakob disease (CJD) which has different types depending on the source. So for those that occur in cattle, they are referred to as Bovine Spongiform Encephalopathy, BSE aka Mad Cow Disease.
This disease can also be named variant Creutzfeldt–Jakob disease (vCJD) because it can affect humans when meat from infected cows containing the diseased PrPsc prion proteins are consumed. Ordinarily, one would expect that, since it is a protein from the meat, the stomach acid should degrade it, well, it cannot be degraded, I believe you already know the reason by now, right?
Another type they cause is the sporadic or classical Creutzfeldt–Jakob disease (cCJD), (sCJD). This is the most common form in humans though rare but occurs.
Sporadic CJD accounts for the greatest number of human deaths from this group of diseases. CJD affects approximately one person per million people each year. So, in a population of 10 million people, there are likely to be at least 10 cases in one year. CJD most often affects people between the ages of 50 to 70 years of age. The cause of sporadic CJD is unknown
The nemesis that befell the Fore people of Papua, New Guinea
After reading the story of this people, I concluded that religious believe and sorcery were the two main factor that led to the death of many of their population, with their women being more on the receiving end of it. Report has it that over 25% of the women in their population died from the prion disease that wrecked havoc in this village.
The Fore people generally believe that when their people die, in order to get to the greater beyond (the land of the dead) to meet "kwelanandamundi", they need to perform some rituals, like keeping food around the body of the dead (so that the good scent of the food will help the dead reach the land of the dead faster and also welcomed), leave the body for 3 days before burying it, and in most cases, they eat the dead body including the brain (Cannibalism).
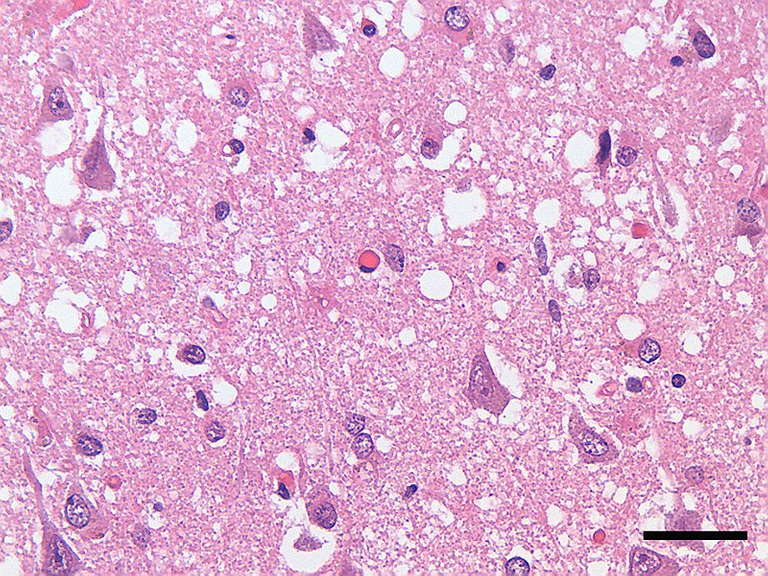
Micrograph showing spongiform degeneration (holes/vacuoles) in the cerebral cortex of a patient who had died of Creutzfeldt-Jakob disease; the scale bar = 30 microns (0.03 mm)
{kind=link}
Prion disease claimed over 1000 lives between 1957 to 1960. An epidemic with a well documented evidence proves that the Fore people got infected with the prion disease through cannibalism. Analysis of the dead victims showed numerous depositions of prion proteins. Just like we earlier explained, they named the disease KURU because of the signs associated with it. People infected with prion disease usually do not live more than a year, and in the case of the sporadic CJD, the patient can even die within six months.
In summary, prion proteins are infectious proteins even though without life, yet that can cause other normal proteins to mis-fold, leading to accumulation of amyloid fibrils in the brain. Prion disease is a neurogenerative disease just like Alzheimer disease caused by the deposition of amyloid-β (Aβ) and Tau protein (MAPT), alpha-synuclein (α-syn/SNCA) in Parkinson disease and Huntingtin (HTT) in Huntingtin disease. They share similar features but prion disease happens to be more deadlier because of its unorthodox means of transmission. Infection is possible through invasive procedures like surgery, especially from surgical equipment that were not well sterilized or initially used on patients with the disease. Even though transmission from human to human through contact has not been well documented, a research has shown that people with sporadic CJD have high concentration of this prion protein in their cornea. Obviously something to be worried about!
If you reached here, you are a great reader, thanks for the read.
References
•The Mystery of Kuru
•What is Scrapie
•1755 and all that: a historical primer of transmissible spongiform encephalopathy
•Prion Protein: The Molecule of Many Forms and Faces
•A brief history of Prions
•Elucidating the function of the prion protein
•Kuru, the First Human Prion Disease
•Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases
•BSE-bovine spongiform encephalopathy
•Creutzfeldt-Jakob disease (CJD)
•William Astbury Conducts the First Studies of Proteins by X-Ray Analysis
•Understanding Kuru
•When People Ate People, A Strange Disease Emerged
•Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients
Thanks.
Welcome back. I was still thinking of you today that it's been a while since we heard from you. Perhaps your real-life engagement got busier.
Happy new year boss. Thanks a bunch.
Yeah, it's really been tight for me, had to first adjust to an entirely new system. Thanks goodness for breaks like this. Period of rebooting...lol
Congratulations!
✅ Good job. Your post has been appreciated and has received support from CHESS BROTHERS ♔ 💪
♟ We invite you to use our hashtag #chessbrothers and learn more about us.
♟♟ You can also reach us on our Discord server and promote your posts there.
♟♟♟ Consider joining our curation trail so we work as a team and you get rewards automatically.
♞♟ Check out our @chessbrotherspro account to learn about the curation process carried out daily by our team.
🥇 If you want to earn profits with your HP delegation and support our project, we invite you to join the Master Investor plan. Here you can learn how to do it.
Kindly
The CHESS BROTHERS team
❤️
Thanks for your contribution to the STEMsocial community. Feel free to join us on discord to get to know the rest of us!
Please consider delegating to the @stemsocial account (85% of the curation rewards are returned).
Thanks for including @stemsocial as a beneficiary, which gives you stronger support.
❤️